Les complications habituelles de la drépanocytose chez l'enfant en Afrique
I. Les premiers signes d'appel
Ce sont essentiellement le syndrome main-pied (Hand Foot Syndrome des anglo-saxons) et l'anémie aiguë que l'on retrouve dans environ la moitié des cas, déjà à partir de l'âge de 6 mois. En Afrique centrale, ces manifestations sont généralement précoces et peuvent même concerner les enfants de moins de 6 mois (2). Le syndrome main-pied est plus rare chez l'enfant drépanocytaire SC.
Il se caractérise par une tuméfaction douloureuse du dos de la main et/ou du pied ; parfois la tuméfaction ne concerne qu'une ou plusieurs phalanges réalisant un tableau de "dactylite" avec des images radiologiques de raréfaction osseuse ou d'ostéocondensation. Par ailleurs, ces tuméfactions peuvent être surinfectées spontanément ou à la suite des tatouages pratiqués lors d'un traitement indigène et peut se compliquer par un abcès ou une ostéomyélite.
Bien que l'anémie aiguë chez le jeune enfant soit souvent liée au paludisme en milieu tropical africain (3), il faut évoquer la possibilité de la drépanocytose même lorsque la goutte épaisse (GE) est positive. Généralement, il s'agit d'une anémie hémolytique, avec une pâleur de la peau, des conjonctives et des muqueuses, souvent extrême, associée ou non à un ictère des bulbes. Les urines sont souvent foncées.
Le taux de l'hémoglobine est généralement bas (< 5g/dl) et le tableau clinique exige souvent une transfusion urgente. Il est prudent dans ces circonstances de prélever une goutte de sang sur papier buvard avant la transfusion afin de ne pas retarder le diagnostic de la drépanocytose chez l'enfant transfusé.
La crise aiguë d'érythroblastopénie, liée à l'infection par un virus particulier, le parvovirus B 19, est une complication grave caractérisée par une anémie progressive et profonde. Elle est très fréquente chez les sujets drépanocytaires et le diagnostic est facilement évoqué devant une anémie sévère accompagnée d'un chiffre de réticulocytes effondré voire nul (4).
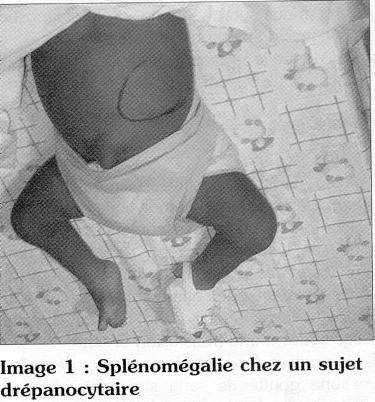
La séquestration splénique et la persistance d'une grosse rate
La brusque augmentation de volume de la rate est une des complications fréquentes observées chez le nourrisson africain et chez le jeune enfant (< 5 ans). C'est un accident imprévisible et qui tue rapidement si elle n'est pas reconnue vite et si le sang n'est pas immédiatement disponible pour la transfusion (5).
Cliniquement, l'enfant présente un état de choc avec une très grosse rate. C'est une situation d'extrême urgence ; c'est pour cela qu'il est très important d'apprendre aux parents à l'identifier en leur apprenant à palper et à connaitre le volume habituel de la rate de leur enfant (Image n°1).
Sur le plan hématologique, on observe une baisse du taux d'Hb par rapport aux valeurs usuelles du patient, une augmentation du taux des réticulocytes et souvent aussi un taux élevé d'érythroblastes circulants.
S'il est généralement admis que la grosse rate est rare chez le drépanocytaire après l'âge de 5 ans (6), il faut signaler qu'elle peut persister au delà de cet âge. En effet, chez le drépanocytaire congolais elle semble persister même au delà de l'adolescence (7) et une observation semblable avait déjà été notée chez les malades nigérians, et la relation possible avec l'endémie palustre avait été évoquée (8). En Afrique subsaharienne, le contexte palustre ne suffit pas pour expliquer cette rate persistante. Elle pourrait être due à un hypersplénisme chronique comme décrit en Jamaïque (9) et chez les drépanocytaires indiens et d'Arabie Saoudite. Ces deux dernières populations se caractérisent aussi par la fréquence élevée des a-thalassémies associées et par le taux élevé de l'hémoglobine foetale (10,11).
II. Les manifestations hémorragiques
Peu décrites en littérature, elles constituent cependant un véritable problème lorsqu'elles se manifestent par des épistaxis torrentiels, parfois dramatiques, justifiant une hospitalisation d'urgence. Dans l'étude faite au Katanga, sur une série de 187 sujets suivis en ambulatoire, on retrouve des antécédents d'épistaxis chez près de la moitié des sujets (46,1 %), avec une fréquence accrue chez le grand enfant et l'adolescent (2). Elles ont été aussi décrites ailleurs en Afrique tropicale (12,13). On peut aussi observer, quoiqu'avec une fréquence moindre, des hémorragies digestives, des gingivorragies et des hématomes du cuir chevelu. Ces complications sont à différencier des manifestations liées à la coagulation intravasculaire disséminée (CIVD) observée au cours des sepsis et infections sévères.
III. Les complications hépatobiliaires
1. Crise douloureuse abdominale
C'est l'une des manifestations les plus fréquentes de la drépanocytose chez l'enfant ; sa cause n'est pas toujours due à une crise vaso-occlusive et d'autres complications doivent alors être évoquées. Parmi ces dernières, il y a l'appendicite aiguë, souvent évoquée à tort et qui conduit parfois à une laparotomie injustifiée.
2. Lithiase biliaire
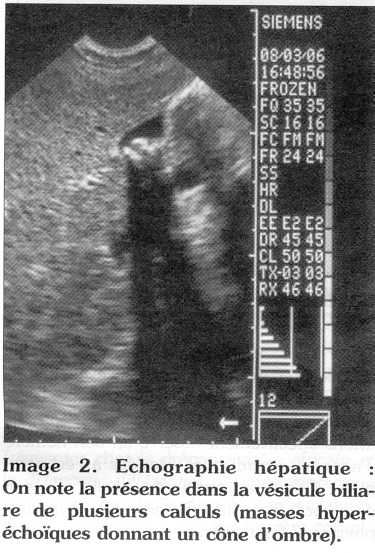
L'autre complication est la lithiase biliaire dont le diagnostic devrait être évoqué, entre autre, en cas d'une exacerbation de l'ictère cutanéo-conjonctival. Sa fréquence chez le drépanocytaire augmente avec l'âge et la sévérité de la maladie ; en Jamaïque, la prévalence est estimée à 12 % dans le groupe d'enfants de 5-7 ans et de 23% dans le groupe âgé de 11-13 ans (14). La lithiase biliaire peut à son tour entraîner d'autres complications : cholécystite aiguë, cholangite et pancréatite aiguë et surtout une septicémie à point de départ biliaire (15). L'examen échographique abdominal devrait être systématique chez le sujet drépanocytaire devant une crise douloureuse abdominale et l'exacerbation de l'ictère. Le diagnostic est évoqué devant une image intravésiculaire, hyperéchoïque et donnant un cône d'ombre (Image 2).
Il semble par ailleurs que l'image de boue biliaire ou "sludgge" soit en faveur d'un stade précoce de lithiase biliaire qui pourrait être cause de pancréatite par obstruction du canal de Wirsung. L'attitude thérapeutique devant une lithiase biliaire asymptomatique (cholécystectomie ou surveillance) chez l'enfant fait encore l'objet de controverse parmi les praticiens.
3. Hépatite virale
L'autre complication hépatobiliaire fréquente chez le drépanocytaire africain est l'hépatite virale, parfois d'origine transfusionnelle. Il faut évoquer ce diagnostic en cas d'aggravation rapide de l'ictère chez l'enfant drépanocytaire présentant ou non une hyperthermie, des douleurs abdominales, des vomissements, une réduction de l'appétit et une profonde asthénie. Les selles peuvent être acholiques. La prévalence des porteurs de l'hépatite B chez les sujets polytransfusés reste encore élevée dans beaucoup de pays d'Afrique tropicale où la sécurité transfusionnelle est insuffisante. La vaccination contre l'hépatite B devrait être obligatoire dans ces groupes à risque.
IV. Les complications ORL et bucco-dentaires
A part les épistaxis, il faut rechercher systématiquement chez l'enfant drépanocytaire la présence d'une hypertrophie des amygdales tonsillaire, de glossite, de mal-occlusion maxillaire et de caries dentaires. Ces anomalies sont fréquentes (16) et devraient justifier une consultation chez le spécialiste ORL et le dentiste.
L'hypertrophie amygdalienne peut contribuer à l'hypoventilation qui contribuerait à provoquer des fréquentes crises vaso-occlusives chez ces patients.
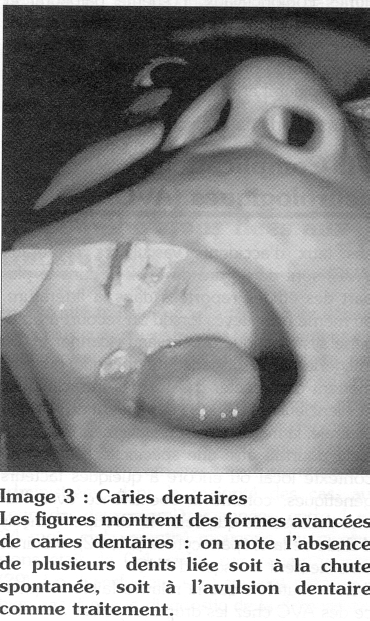
Les caries dentaires sont très fréquentes chez l'enfant drépanocytaire (Image 3) ; dans notre expérience, elles concernent près de 60 % de sujets homozygotes SS, dès l'âge de 3 ans (2). Ces caries témoignent, au-delà d'une mauvaise hygiène bucco-dentaire, d'une fragilité particulière à rattacher probablement à la maladie drépanocytaire. L'éducation à la santé en matière d'hygiène buccale et les mesures préventives des caries dentaires méritent une attention particulière dans le suivi de l'enfant drépanocytaire africain.
V. Complications oculaires
Les complications habituelles sont les rétinopathies dont le dépistage systématique est indiqué à partir de l'adolescence. Elles sont souvent caractérisées par la tortuosité et la dilatation des vaisseaux rétiniens ; les hémorragies sont relativement rares (17).
En dehors de ces lésions, il faut signaler les signes conjonctivaux, l'oedème palpébral et une exophtalmie qui peut être mono ou bilatérale vraisemblablement liée à l'infarctus osseux fronto-orbitaire ou encore à un hématome épidural et rétro-orbitaire (18).
VI. Complications neurologiques (AVC)
Les taux d'accidents vasculaires cérébraux (AVC) sont de l'ordre de 5-12 % dans la plupart des études reportées dans la littérature, ou même plus élevés lorsqu'on recourt à l'imagerie systématique du cerveau (scanner et/ou imagerie par résonnance magnétique) qui permettent de détecter des formes infracliniques et silencieuses (19, 20) ; cependant en Afrique centrale, la fréquence est relativement faible (2). Cela pourrait être une spécificité liée soit au contexte local ou encore à quelques facteurs génétiques, comme la coexistence avec l'alpha-thalassémie qui, selon le Groupe Français d'Eude de la drépanocytose (21, 22), réduirait sensiblement le risque d'AVC.
Il note toutefois que la relative faible fréquence des AVC chez les drépanocytaires africains pourrait être liée à une sous-estimation des formes infracliniques ou frustres qui passeraient inaperçues par manque de moyens d'investigations appropriées. L'étude systématique du flux sanguin des artères de la base du crâne par doppler transcrânien pourrait être envisageable actuellement dans plusieurs pays africains.
VII. Les complications ostéo-articulaires
Contrairement aux AVC, les complications ostéo-articulaires sont particulièrement fréquentes dans notre population. Elles sont dominées par les ostéomyélites et les nécroses aseptiques de la hanche. Les ostéomyélites sont fréquentes chez le jeune enfant et peuvent intéresser plusieurs membres à la fois ; elles sont souvent fistulisées et rebelles au traitement antibiotique. Les micro-organismes les plus incriminés sont Salmonella spp et Staphylococcus aureus (23).
Les nécroses aseptiques de la hanche ont une incidence variable selon les populations, l'âge et les moyens d'explorations : la fréquence est de 9,2 % au Sénégal (24), 13,7 % au Nigéria (25) et de 5 % en Inde (26). Elles sont relativement rares chez le jeune enfant et se retrouvent surtout dans la tranche d'âge de 14-25 ans (25,27) ; il faut donc savoir les rechercher systématiquement chez le grand enfant et surtout chez l'adolescent qui présente une douleur de hanche ou une boiterie.
VIII. Pathologies cardiovasculaires
La présence d'un souffle cardiaque râpeux chez le drépanocytaire doit fait évoquer une cause autre que l'anémie. Dans une cohorte d'environ 300 drépanocytaires suivis, nous avions effectivement observé un bon nombre d'anomalies cardiaques décelées à l'échographie ; il s'agissait surtout de valvulopathies et de myocardiopathies dilatées et ou hypertrophiques. (Observation personnelle).
Le suivi cardiovasculaire des patients drépanocytaires doit donc comporter un bilan échocardiographique régulier.
IX. Pathologies infectieuses
Les infections sont responsables d'une part importante de la mortalité et de la morbidité de la drépanocytose.
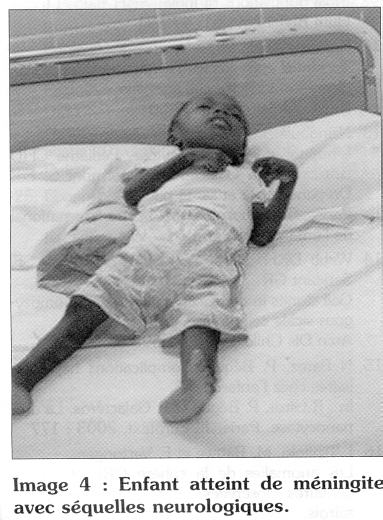
Les méningites et les septicémies sont les infections les plus graves avec un pronostic souvent réservé en milieu tropical et souvent accompagnées de séquelles sévères (Image 4). Les germes responsables sont surtout le Streptococcus pneumoniae, l'Haemophilus influenzae et la Samonella sp.
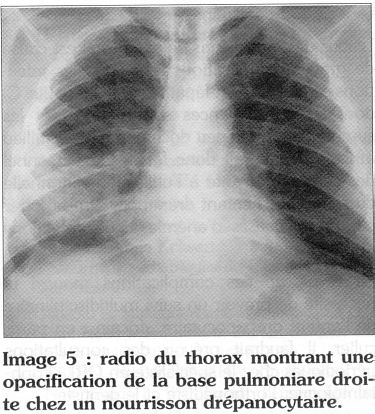
Les infections pulmonaires sont particulièrement sévères et mortelles chez les nourrissons qui, pour la plupart, vont présenter un tableau de pneumonie massive (Image 5) avant même que le diagnostic de drépanocytose n'ait été évoqué. Les infections pulmonaires sont fréquentes chez le drépanocytaire africain et le diagnostic différentiel de crise thoracique aiguë n'est pas facile dans la pratique quotidienne; ce qui fait que cette dernière complication n'est que rarement retenue chez les drépanocytaires de moins de 15 ans.
Signalons d'autre part, la fréquence non négligeable de la tuberculose, tout particulièrement dans sa manifestation osseuse. Il faut savoir l'évoquer en cas de pneumopathie traînante chez un enfant drépanocytaire ou en cas de déformation de la colonne vertébrale.
Nous avons, en effet, dans une série de 137 enfants tuberculeux, répertorié 9 cas de maladie de Pott dont 6 chez des enfants drépanocytaires (28).
X. Complications liées aux transfusions
La sécurité transfusionnelle fait encore défaut dans plusieurs pays africains, spécialement en milieu rural. Or le sujet drépanocytaire est soumis à plusieurs transfusions, parfois d'une manière abusive. Ainsi, par exemple, dans une série de 186 drépanocytaires régulièrement suivis au Katanga, 80% de sujets étaient transfusés. Les premières transfusions étaient administrées, dans la majorité des cas, au cours des deux premières années, entre 4 et 24 mois, et la quantité moyenne de transfusions était de 0.4 unités par patient/année (29). Les transfusions sont souvent précoces, bien avant que le diagnostic de la drépanocytose ne soit déterminé.
L'abus de transfusions de sang total est cause de graves complications telles que les infections virales (HIV, Hépatite B et Hépatite C) dont les conséquences sont néfastes et les surcharges en fer peu documentées en milieu africain. Il faudrait donc former le personnel des centres de santé à l'utilisation rationnelle du sang chez l'enfant drépanocytaire et lutter contre les causes d'anémie aiguë (infections et paludisme).
La revue de ces complications montre la nécessité de prévoir un suivi multidisciplinaire de l'enfant drépanocytaire africain : en particulier, il faudrait prévoir des consultations périodiques chez le spécialiste en ORL, l'ophtalmologue, l'orthopédiste et le dentiste.
Références
- Angastiniotis, B. Modell, P.Englezos and B. Boulyjenkov. Prevention and control of haemoglobinopathies Bull WHO 1995, 73 (3): 375-386
- Tshilolo, R. Mukendi and R. Girot. La drépanocytose dans le sud du Zaire. Etude de deux séries de 251 et 340 malades suivis entre 1988 et 1992. Arch Pédiatr 1996 ; 3: 101-3
- Newton C.R, Warn P.A, Winstanley P.A, Severe anemia living in children in a malaria endemic area of Kenya. Trop Med and Intern Health 1997, 2(2): 165-178
- Palie C, M Leruez Ville, A Cécille, I Vassias, J. Le Junter et F. Morinet. Le parvovirus B et l'hématopoïèse Hématologie 1995; 1(6) : 461-468
- Edmond AM, Collis R, Darvill D, Higgs DR, Maude GH, Serjeant GR
Acute splenic sequestration in homozygous sickle cell disease : natural history and management. J Pediatr 1985: 107: 201-6 - Begue P, Assimadi K. Diagnostic de la drépanocytose et de ses complications.
In : La Maladie drépanocytaire - Ed. Labo Sandoz, 1984, 78. - Tshilolo M, Mukendi K et Wembonyama O. Analyse des problèmes pathologiques et médicosociaux de l'enfant drépanocytaire zaïrois. "Expérience de l'hôpital Gécamines de Kolwezi". Pédiatrie en Afrique 1993 ; 12: 25-28
- Adekile AD, Mckie KM, Adeodu O et al. Persistent gross splenomegaly in Nigerian patients with sickle cell anaemia: relationship to malaria. Ann Trop Paediatr 1988 ; 8: 103-7
- Serjeant, GR. The clinical features of sickle cell disease. In Higgs DR, Weatherall DJ, eds. The haemoglobinopathies. Londres : Baillère Tindall, 1993 ; 93-115
- Miller BA, Salameh M, Ahmed M et al.High fetal hemoglobin production in sickle cell anemia in the Eastern province of Saudi Arabia is genetically determined. Blood 1986; 67: 1404-1
- Labie D, Srinivas R, Dunda O et al Haplotypes in tribal Indians bearing the sickle gene: evidence for the unicentric origin of the BS mutation and the unicentric origin of the tribal populations of India. Hum Biol 1989; 61: 479-91
- Konotey-Ahulu FID. Torrential epistaxis with symmetrical facial skin ulceration in sickle cell anaemia. Br Med J 1965; 2: 859-60
- Nzingoula S. L'hopital et la drépanocytose. Pédiatrie CHU Brazzaville. In Galacteros F, Dormont S. Eds Drépanocytose et santé publique. Paris : Co-édition Inserm/Centre International de l'enfance, 1991 : 161-71
- Webb DKH, Darby JS, Dunn DT, Terry SF, Serjeant GR. Gall stones in Jamaican children with homozygous sickle cell disease. Arch Dis Child 1989; 64:693-6.
- N Parez, P. Bégué. Complications hépatobiliaires chez l'enfant.
In : R.Girot, P. Bégué et F. Galactéros. La drépanocytose. Paris, JL Eurotext, 2003 : 177 - L.Tshilolo, M. Bahwe et F. Vertongen. Les anomalies de la sphère ORL et buccodentaires observées chez le drépanocytaire zairois. Nouv Rev Fr Hematol 1995 ; 37(1) : 4( Abstract 11)
- Kaimbo K, Ngiyulu M, Dralands et al. Ocular findings in children with homozygous sickle cell disease in the Democratic Republic of Congo. Bull Soc Belge Ophtalmol 2000; 275: 27-30
- Mallouh A, Young M, Hamdam J et Salamah M: Proptosis, skull infarction, and retro-orbital and epidural hematomas in a child with sickle cell disease. Clin Pediatr 1987, 26, 536-8
- Hoppe C, Klitz W, Cheng S et al. Gene interactions and stroke risk in children with sickle cell anemia. Blood.2003 Nov 13:
- Manci EA, Culberson DE, Yang YM et al Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003; 123(2): 359-65
- Neonato MG, Guilloud-Bataille M, Beauvais P et al. Acute clinical events in 299 homozygous sickle cell patients living in France. French Study Group on Sickle Cell Disease. Eur J Haematol. 2000; 65(3): 155-64
- Hsu LL, Miller ST, Wright E, et al. Alpha Thalassemia associated with decreased risk of abnormal transcranial Doppler ultrasonography in children with sickle cell anemia. J Pediatr Hematol Onvcol. 2003; 25(8):622-8
- Burnet MW, Bass JW et Cook BA. Etiology of osteomyelitis complicating sickle cell disease. Pediatrics, 1998, 101, 296-7
- Diop S, Mokono SO, Ndiaye M et al Homozygous sickle cell disease in patients above 20 years of follow-up of 108 patients in Dakar. Rev Med Interne, 2003; 24(11):711-5
- Ebong WW. Avascular necrosis of the femoral head associated with haemoglobinopathy.
Trop Geogr Med 1977, 29: 19-23 - Hawker H, Neilson H, Hayes RJ et Serjeant GR Haemaotological factors associated with avascular necrosis of the femoral head in homozygous sickle cell disease. Br J Haematol, 1982; 50: 29-34
- Ndugwa CM. Aseptic necrosis of the head of the femur among sickle cell anaemia patients in Uganda East Afr Med j 1992 ; 69(10): 572-6
- L.Tshilolo et Mukendi R, La tuberculose chez l'enfant. Notre expérience à l'HP GCM de Kolwezi. I° Journées pédiatriques du Shaba, Lubumbashi, Zaire 1989.
- Tshilolo L, Mukendi R and Wembonyama O Total blood transfusion in Zairian children and young adults with sickle cell disease INSERM/NIH/WHO/EHA Conference on "New trends in therapy for hemoglobinopathies and thalassemias". Paris, September 19-22, 1994.
Développement et Santé, n°182, 2006